力场(化学)
编辑在化学和分子建模的背景下,力场是一种计算方法,用于估计分子内原子之间以及分子之间的力。更准确地说,力场是指用于计算分子力学、分子动力学或蒙特卡罗模拟中原子或粗粒粒子系统的势能的函数形式和参数集。
所选能量函数的参数可以来自物理和化学实验、量子力学计算,或两者兼而有之。
力场是原子间势,使用与经典物理学中的力场相同的概念,不同之处在于化学中的力场参数描述了能量景观,全原子力场为系统中每种类型的原子(包括氢)提供参数,而联合原子间势将甲基和亚甲基桥中的氢和碳原子视为一个相互作用中心。粗粒度势能通常用于长时间模拟蛋白质、核酸和多组分复合物等大分子,但会牺牲化学细节以获得更高的计算效率。
力场功能形式
编辑分子力学中势能的基本函数形式包括通过共价键连接的原子相互作用的键合项,以及描述长程静电力和范德华力的非键合(也称为非共价)项。项的具体分解取决于力场,但加性力场中总能量的一般形式可以写为乙全部的=乙保税+乙非保税{displaystyleE_{text{total}}=E_{text{bonded}}+E_{text{nonbonded}}}其中共价和非共价贡献的成分由以下总和给出:乙保税=乙纽带+乙角度乙非保税=乙静电+乙范德华键和角项通常由不允许键断裂的二次能量函数建模。
更昂贵的莫尔斯电势提供了对更高拉伸共价键更真实的描述。二面体能量的函数形式从一个力场到另一个力场是可变的。此外,可能会添加不适当的扭转项以加强芳环和其他共轭系统的平面性,以及描述不同内部变量(例如角度和键长)耦合的交叉项。一些力场还包括氢键的明确术语。非键项在计算上是最密集的。
一个流行的选择是将相互作用限制为成对能量。范德华项通常用Lennard-Jones势计算,静电项用库仑定律计算。然而,两者都可以通过一个常数因子进行缓冲或缩放,以说明电子极化率。
自1970年代以来,具有这种能量表达的研究一直集中在生物分子上,并在2000年代初期推广到元素周期表中的化合物,包括金属、陶瓷、矿物和有机化合物。确定分子动力学模拟中的振动频率。原子之间的键越强,力常数的值就越高,IR/拉曼光谱中的波数(能量)就越高。
根据给定的力常数的振动谱可以从具有~1fs时间步长的短MD轨迹(5ps)、速度自相关函数的计算及其傅里叶变换来计算。尽管胡克定律的公式在平衡距离附近的键长处提供了合理的准确度,但随着距离的移动,它的准确度就会降低。为了更好地模拟莫尔斯曲线,可以使用三次和更高的幂。
然而,对于大多数实际应用来说,这些差异可以忽略不计,并且键长预测的不准确性约为千分之一埃,这也是常见力场可靠性的限制。可以改为使用莫尔斯势来实现键断裂和更高的准确性,即使它的计算效率较低。原子电荷可以对势能做出主要贡献,特别是对于极性分子和离子化合物,并且对于模拟几何形状、相互作用能以及反应性至关重要。
原子电荷的分配通常仍然遵循经验和不可靠的量子力学协议,这通常导致相对于与实验偶极矩和理论一致的物理合理值的几个xxx的不确定性。基于电子变形密度、内部偶极矩和扩展玻恩模型的实验数据,已开发出可再现的力场原子电荷。不确定性<10%,或±0.1e,
参数化
编辑除了势的函数形式外,力场还为不同类型的原子、化学键、二面角、平面外相互作用、非键相互作用和可能的其他术语定义了一组参数。许多参数集是经验性的,一些力场使用难以分配物理解释的广泛拟合项。
原子类型是为不同的元素以及在足够不同的化学环境中的相同元素定义的。例如,水中的氧原子和羰基官能团中的氧原子被分类为不同的力场类型。典型的力场参数集包括原子质量、原子电荷、每种原子类型的Lennard-Jones参数值,以及键长、键角和二面角的平衡值。键合术语是指键合原子对、三联体和四联体,包括每个势能的有效弹簧常数值。
大多数当前的力场参数使用固定电荷模型,通过该模型,每个原子都被分配一个不受局部静电环境影响的原子电荷值。具有最 大精度和可传输性的模拟的力场参数化,例如IFF,遵循一个明确定义的协议。
该工作流程可能涉及(1)检索X射线晶体结构或化学式,(2)定义原子类型,(3)获得原子电荷,(4)分配初始Lennard-Jones和键合参数,(5)相对于实验参考数据的密度和几何形状,(6)相对于实验参考数据的能量特性(表面能、水合能)的计算测试,(7)二次验证和细化(热、机械和扩散特性)。
主要的迭代循环发生在步骤(5)和(4)之间,以及(6)和(4)/(3)之间。参数的化学解释和可靠的实验参考数据起着至关重要的作用。蛋白质、DNA和RNA等生物大分子的分子模拟参数通常来自对有机小分子的观察,这些参数更容易用于实验研究和量子计算。
因此,出现了多个问题,例如
(1)来自量子计算的不可靠的原子电荷可能会影响所有计算的性质和内部一致性,
(2)来自气相分子的不同量子力学的数据可能无法用于凝聚态分子的模拟阶段,
(3)小分子数据的使用和对较大聚合物结构的应用涉及不确定性,
(4)不同的实验数据在准确性和参考状态(例如温度)方面的变化可能会导致偏差。
因此,已经报道了生物分子的发散力场参数。实验参考数据包括例如汽化焓(OPLS)、升华焓、偶极矩和各种光谱参数。通过解释所有力场参数并选择一致的参考状态(例如室温和大气压),可以克服不一致的情况。
一些力场还包括没有明确的化学原理、参数化协议、关键特性(结构和能量)的不完整验证、缺乏对参数的解释以及对不确定性的讨论。在这些情况下,已经报告了计算属性的大的随机偏差。
方法
一些力场包括极化率的明确模型,其中粒子的有效电荷可能受到与其邻居的静电相互作用的影响。核壳模型很常见,它由一个带正电的核心粒子(代表可极化原子)和一个通过弹簧状谐振子电位附着在核心原子上的带负电粒子组成。
最近的例子包括具有虚拟电子的可极化模型,可再现金属中的图像电荷和可极化的生物分子力场。通过为极化率增加这样的自由度,参数的解释变得更加困难,并增加了任意拟合参数的风险和降低的兼容性。
由于需要重复计算局部静电场,计算费用增加。可极化模型在捕获基本化学特征并且净原子电荷相对准确(±10%以内)时表现良好。最近,此类模型被错误地称为DrudeOscillator势。
这些模型的适当术语是洛伦兹振荡器模型,因为洛伦兹而不是德鲁德提出了某种形式的电子与原子核的连接。Drude模型假设电子的运动不受限制,例如金属中的自由电子气。
参数化
从历史上看,已经采用了许多方法来参数化力场。许多经典力场依赖于相对不透明的参数化协议,例如,使用近似的量子力学计算,通常在气相中,期望与凝聚相特性和电位的经验修改以匹配实验观测值的一些相关性。
协议可能不可重复,半自动化通常在生成参数、优化参数生成和广泛覆盖方面发挥作用,而不是化学一致性、可解释性、可靠性和可持续性。
类似地,最近甚至可以使用更多自动化工具来参数化新的力场,并帮助用户为迄今为止尚未参数化的化学物质开发自己的参数集。努力提供开源代码和方法的有openMM和openMD。
在没有化学知识输入的情况下使用半自动化或全自动化可能会增加原子电荷水平的不一致性,用于分配剩余参数,并可能削弱参数的可解释性和性能。
界面力场(IFF)对整个周期内的所有化合物假设一个单一的能量表达式(具有9-6和12-6LJ选项),并使用标准化模拟协议进行严格验证,从而实现参数的完全可解释性和兼容性,以及准确度高并且可以访问无限的化合物组合。
可转移性
编辑功能形式和参数集已由原子间势的开发人员定义,并具有可变程度的自洽性和可转移性。当势项的函数形式不同时,一个原子间势函数的参数通常不能与另一个原子间势函数一起使用。在某些情况下,可以通过较小的努力进行修改,例如,从9-6Lennard-Jones势到12-6Lennard-Jones势。相反,从Buckingham势到谐波势,或从嵌入式原子模型到谐波势的转换需要许多额外的假设并且可能是不可能的。
限制
编辑所有原子间势均基于近似值和实验数据,因此通常称为经验势。性能变化范围从比密度泛函理论计算更高的精度,可以访问百万倍大的系统和时间尺度,到根据力场的随机猜测。与DFT级量子方法相比,使用化学键的准确表示,结合可重复的实验数据和验证,可以以更少的参数和假设产生持久的高质量原子间势。可能的限制包括原子电荷,也称为点电荷。
大多数力场依靠点电荷来重现分子周围的静电势,这对于各向异性电荷分布的效果较差。补救措施是点电荷有一个清晰的解释,并且可以添加虚拟电子来捕获电子结构的基本特征,例如金属系统中额外的极化率来描述图像电位,π共轭系统中的内部多极矩和孤对在水里。通过使用可极化力场或使用宏观介电常数可以更好地包括环境的电子极化。然而,一个介电常数值的应用是在蛋白质、生物膜、矿物质、所有类型的范德华力也强烈依赖于环境,因为这些力源自诱导偶极子和瞬时偶极子的相互作用(参见分子间力)。这些力的原始弗里茨伦敦理论仅适用于真空。
ADMcLachlan于1963年提出了一种更一般的关于压缩介质中范德华力的理论,并将最初的伦敦方法作为一个特例。麦克拉克伦理论预测,介质中的范德华引力比真空中的弱,并且遵循同类溶解的规律,这意味着不同类型的原子比相同类型的原子相互作用更弱。这与用于发展经典力场的组合规则或Slater-Kirkwood方程形成对比。组合规则表明,两个不同原子(例如,C...N)的相互作用能是相应相同原子对(即,C...C和N...N)的相互作用能的平均值。根据麦克拉克伦的理论,介质中粒子的相互作用甚至可以是完全排斥的,正如液氦所观察到的那样,然而,缺乏汽化和冰点的存在与纯粹排斥相互作用的理论相矛盾。
JacobIsraelachvili已经解释了不同材料之间的吸引力(Hamaker常数)的测量。例如,烃类在水中的相互作用约为真空中的10%。在蛋白质结构细化方面,人们强烈感受到了局限性。主要的潜在挑战是聚合物分子的巨大构象空间,当包含超过20个单体时,它会超出当前的计算可行性。蛋白质结构预测(CASP)关键评估的参与者并没有试图改进他们的模型以避免分子力学的中心尴尬,即能量最小化或分子动力学通常会导致模型不像实验结构。力场已成功应用于不同X射线晶体学和NMR光谱学应用中的蛋白质结构细化,尤其是使用XPLOR程序。
然而,改进主要由一组实验约束驱动,原子间势主要用于消除原子间障碍。计算结果实际上与在程序DYANA中实现的刚性球势(从NMR数据计算)或完全不使用能量函数的晶体学细化程序相同。这些缺点与原子间势和无法有效地对大分子的构象空间进行采样有关。因此,解决此类大规模问题的参数开发也需要新的方法。一个特定的问题领域是蛋白质的同源性建模。同时,已经开发了用于配体对接、蛋白质折叠、同源模型改进、计算蛋白质设计、还有人认为,一些蛋白质力场以与蛋白质折叠或配体结合无关的能量运行。蛋白质力场的参数再现了升华焓,即分子晶体的蒸发能量。
然而,蛋白质折叠和配体结合在热力学上更接近于结晶或液固转变,因为这些过程代表了凝聚介质中移动分子的冻结。因此,预计蛋白质折叠或配体结合过程中的自由能变化代表类似于融合热(分子晶体熔化过程中吸收的能量)的能量、构象熵贡献和溶剂化自由能的组合。熔化热明显小于升华焓。
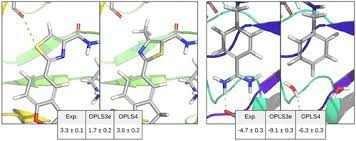
因此,描述蛋白质折叠或配体结合的潜力需要更一致的参数化协议,例如,如IFF所述。实际上,当从蛋白质工程或α螺旋到线圈跃迁数据估计时,蛋白质中H键的能量约为-1.5kcal/mol,但从分子晶体的升华焓估计的相同能量为-4到-6kcal/mol,这与重新形成现有的氢键和不从头开始形成氢键有关。正如McLachlan理论所预测的,由蛋白质工程数据得出的修改后的Lennard-Jones势的深度也小于典型的势参数,并遵循类似溶解的规则。
从蛋白质工程或α螺旋到线圈跃迁数据估计为5kcal/mol,但从分子晶体的升华焓估计的相同能量为-4到-6kcal/mol,这与重新形成现有氢键有关,而不是从头开始形成氢键。
正如McLachlan理论所预测的,由蛋白质工程数据得出的修改后的Lennard-Jones势的深度也小于典型的势参数,并遵循类似溶解的规则。
从蛋白质工程或α螺旋到线圈跃迁数据估计为5kcal/mol,但从分子晶体的升华焓估计的相同能量为-4到-6kcal/mol,这与重新形成现有氢键有关,而不是从头开始形成氢键。
正如McLachlan理论所预测的,由蛋白质工程数据得出的修改后的Lennard-Jones势的深度也小于典型的势参数,并遵循类似溶解的规则。
广泛使用的力场
编辑不同的力场是为不同的目的而设计的。所有这些都在各种计算机软件中实现。MM2由NormanAllinger开发,主要用于碳氢化合物和其他有机小分子的构象分析。它旨在尽可能精确地再现分子的平衡共价几何形状。
它实现了大量参数,这些参数针对许多不同类别的有机化合物(MM3和MM4)不断改进和更新。CFF由AriehWarshel、Lifson和同事开发,作为统一研究一般分子和分子晶体的能量、结构和振动的通用方法。
由Levitt和Warshel开发的CFF程序基于所有原子的笛卡尔表示,它是许多后续模拟程序的基础。ECEPP专为肽和蛋白质的建模而开发。它使用氨基酸残基的固定几何形状来简化势能面。因此,能量最小化是在蛋白质扭转角的空间中进行的。
MM2和ECEPP都包括H键势能和用于描述围绕单键旋转的扭转势能。ECEPP/3在InternalCoordinateMechanics和FANTOM中实施(有一些修改)。AMBER、CHARMM和GROMOS主要用于大分子的分子动力学,尽管它们也常用于能量最小化。
因此,所有原子的坐标都被视为自由变量。界面力场(IFF)被开发为元素周期表中化合物的第 一个一致力场。
它克服了分配一致电荷的已知限制,利用标准条件作为参考状态,再现结构、能量和能量衍生物,并量化所有包含化合物的限制。它兼容多种力场来模拟混合材料(CHARMM、AMBER、OPLS-AA、CFF、CVFF、GROMOS)。
古典
- AMBER(辅助模型构建和能量优化)——广泛用于蛋白质和DNA。
- CFF(一致力场)——适用于各种有机化合物的一系列力场,包括聚合物、金属等的力场。
- CHARMM(哈佛分子力学化学)——最初在哈佛开发,广泛用于小分子和大分子
- COSMOS-NMR–适用于各种无机化合物、有机化合物和生物大分子的混合QM/MM力场,包括原子电荷NMR特性的半经验计算。COSMOS-NMR针对基于NMR的结构解析进行了优化,并在COSMOS分子建模包中实现。
- CVFF–也广泛用于小分子和大分子。
- ECEPP——多肽分子的xxx个力场——由FAMomany、HAScheraga及其同事开发。
- GROMOS(格罗宁根分子模拟)——作为GROMOS软件一部分的力场,这是一种用于研究生物分子系统的通用分子动力学计算机模拟软件包。GROMOS力场A版已开发用于蛋白质、核苷酸和糖的水溶液或非极性溶液。还提供用于模拟气相分离分子的B版本。
- IFF(界面力场)——xxx个在一个平台上覆盖金属、矿物、2D材料和聚合物的力场,具有尖端的精度并与许多其他力场(CHARMM、AMBER、OPLS-AA、CFF、CVFF、GROMOS)兼容),包括12-6LJ和9-6LJ选项
- MMFF(默克分子力场)——由默克公司为广泛的分子开发。
- OPLS(液体模拟的优化潜力)(变体包括OPLS-AA、OPLS-UA、OPLS-2001、OPLS-2005、OPLS3e、OPLS4)——由耶鲁大学化学系的WilliamL.Jorgensen开发。
- QCFF/PI–共轭分子的一般力场。
- UFF(通用力场)–一种通用力场,其参数适用于包括锕系元素在内的完整元素周期表,由科罗拉多州立大学开发。由于缺乏对几乎所有要求保护的化合物(尤其是金属和无机化合物)的参数的验证和解释,可靠性很差。
可极化
- AMBER–JimCaldwell及其同事开发的极化力场。
- AMOEBA(用于生物分子应用的原子多极优化能量学)——由PengyuRen(德克萨斯大学奥斯汀分校)和JayW.Ponder(华盛顿大学)开发的力场。AMOEBA力场正在逐渐向物理更丰富的AMOEBA+移动。
- CHARMM–由S.Patel(特拉华大学)和CLBrooksIII(密歇根大学)开发的极化力场。基于A.MacKerell(马里兰大学巴尔的摩分校)和B.Roux(芝加哥大学)开发的经典Drude振荡器。
- CFF/ind和ENZYMIX–xxx个极化力场,随后被用于生物系统的许多应用中。
- COSMOS-NMR(分子结构的计算机模拟)——由UlrichSternberg及其同事开发。混合QM/MM力场能够使用具有快速BPT形式的局部键轨道对静电特性进行显式量子力学计算。在每个分子动力学步骤中,原子电荷波动都是可能的。
- P.Th.开发的DRF90。vanDuijnen和同事。
- IFF(界面力场)——包括金属(Au、W)和pi共轭分子的极化率
- NEMO(非经验分子轨道)——由GunnarKarlström和隆德大学(瑞典)的同事开发的程序
- PIPF–流体的可极化分子间势是有机液体和生物聚合物的诱导点偶极力场。分子极化是基于Thole的相互作用偶极子(TID)模型,由嘉利高高研究组开发|在明尼苏达大学。
- 极化力场(PFF)–由RichardA.Friesner及其同事开发。
- 基于SP的化学势均衡(CPE)–由R.Chelli和P.Procacci开发的方法。
- PHAST-由ChrisCioce和同事开发的极化电位。
- ORIENT–由AnthonyJ.Stone(剑桥大学)和同事开发的程序。
- 高斯静电模型(GEM)——一种基于密度拟合的极化力场,由NIEHS的ThomasA.Darden和G.AndrésCisneros开发;和巴黎第六大学的Jean-PhilipPiquemal。
- 液体、电解质和聚合物的原子极化电位(APPLE&P),由OlegBorogin、DmitryBedrov和同事开发,由WasatchMolecularIncorporated发行。
- 基于由JürgHutter及其同事(苏黎世大学)开发的Kim-Gordon方法的极化程序
- GFN-FF(几何、频率和非共价相互作用力场)——由大学的StefanGrimme和SebastianSpicher开发的完全自动化的部分极化通用力场,用于准确描述元素周期表中大分子的结构和动力学波恩。
反应性
- EVB(经验价键)——由Warshel及其同事引入的这种反应力场可能是在不同环境中使用力场模拟化学反应的最可靠和物理上一致的方法。EVB有助于计算凝聚相和酶中的活化自由能。
- ReaxFF–由AdrivanDuin、WilliamGoddard和同事开发的反应力场(原子间势)。它比经典的MD(50x)慢,需要经过特定验证的参数集,并且没有对表面和界面能的验证。参数是不可解释的。它可以用于化学反应的原子级动力学模拟。并行化ReaxFF允许在大型超级计算机上对>>1,000,000个原子进行反应模拟。
粗粒度
- DPD(耗散粒子动力学)-这是化学工程中常用的一种方法。它通常用于研究各种简单和复杂流体的流体动力学,这些流体需要考虑比经典分子动力学更容易获得的时间和长度尺度。潜力最初是由Hoogerbrugge和Koelman提出的,后来由Español和Warren进行了修改。2008年的CECAM研讨会上很好地记录了当前的技术状态。最近,已经着手捕捉一些与解决方案相关的化学字幕。这导致了考虑将DPD相互作用势与实验可观察物进行自动参数化的工作。
- MARTINI——由格罗宁根大学的Marrink和同事开发的粗粒度电位,最初是为脂质的分子动力学模拟而开发的,后来扩展到各种其他分子。力场将四个重原子映射到一个CG相互作用位点,并进行参数化以再现热力学性质。
- SAFT-伦敦帝国理工学院分子系统工程组开发的一种自上而下的粗粒度模型,使用SAFT状态方程拟合纯化合物的液相密度和蒸气压。
- SIRAH–由Pantano和乌拉圭蒙得维的亚巴斯德研究所生物分子模拟小组的同事开发的粗粒度力场;开发用于水、DNA和蛋白质的分子动力学。AMBER和GROMACS软件包免费提供。
- VAMM(虚拟原子分子力学)——由Korkut和Hendrickson开发的粗粒度力场,用于分子力学计算,例如基于C-α原子的虚拟相互作用的大规模构象转变。它是一个基于知识的力场,用于捕获依赖于蛋白质二级结构和残基特定接触信息的特征。
机器学习
- ANI(ArtificialNarrowIntelligence)是一种可转移的神经网络势,由原子环境向量构建而成,能够提供能量方面的DFT精度。
- FFLUX(最初是QCTFF)一组经过训练的克里金模型,它们一起运行以提供在分子中的原子或量子化学拓扑能量项(包括静电、交换和电子相关)上训练的分子力场。
- TensorMol,一个混合模型,一个神经网络提供了一个短程潜力,而更传统的潜力增加了筛选的长期项。
- Δ-ML不是一种力场方法,而是一种模型,该模型将学习的校正能量项添加到近似且计算上相对便宜的量子化学方法,以提供更高阶、计算上更昂贵的量子化学模型的准确度水平。
- SchNet一个利用连续滤波器卷积层的神经网络,用于预测化学性质和势能表面。
- PhysNet是一种基于神经网络的能量函数,用于预测能量、力和(波动的)部分电荷。
水
用于模拟水或水溶液(基本上是水的力场)的一组参数称为水模型。水因其不寻常的特性和作为溶剂的重要性而引起了广泛的关注。已经提出了许多水模型;一些例子是TIP3P、TIP4P、SPC、灵活的简单点充电水模型(flexibleSPC)、ST2和mW。其他溶剂和溶剂表示方法也应用于计算化学和物理学中,一些示例在溶剂模型页面上给出。最近,已经发表了用于生成水模型的新方法。
修饰氨基酸
- Forcefield_PTM–基于AMBER的力场和网络工具,用于对ChrisFloudas及其同事开发的蛋白质中氨基酸的常见翻译后修饰进行建模。它使用ff03电荷模型,并有几个侧链扭转校正参数化以匹配量子化学旋转表面。
- Forcefield_NCAA-基于AMBER的力场和网络工具,用于使用ff03电荷模型在凝聚相模拟中对蛋白质中的常见非天然氨基酸进行建模。据报道,这些电荷与相应侧链类似物的水合自由能相关。
其他
- LFMM(配体场分子力学)-基于角重叠模型(AOM)的过渡金属周围配位球的函数。在分子操作环境(MOE)中作为DommiMOE和Tinker实施
- VALBOND-一种基于价键理论的角度弯曲函数,适用于大角度扭曲、高价分子和过渡金属配合物。它可以并入其他力场,例如CHARMM和UFF。
内容由匿名用户提供,本内容不代表vibaike.com立场,内容投诉举报请联系vibaike.com客服。如若转载,请注明出处:https://vibaike.com/151252/