哈特里-福克方程简介
编辑在计算物理和化学中,Hartree–Fock (HF) 方法是一种用于确定定态量子多体系统的波函数和能量的近似方法。
哈特里-福克方法通常假设系统的精确 N 体波函数可以用单个斯莱特行列式(在粒子是费米子的情况下)或单个xxx(在玻色子的情况下)来近似 ) 的 N 个自旋轨道。 通过调用变分法,可以推导出一组 N 个自旋轨道的 N 耦合方程。 这些方程的解产生 Hartree–Fock 波函数和系统的能量。
特别是在较早的文献中,哈特里-福克方程序也被称为自洽场法(SCF)。 在推导出现在称为 Hartree 方程(作为薛定谔方程的近似解)的过程中,Hartree 要求根据电荷分布计算的最终场与假设的初始场自洽。 因此,自洽性是解决方案的要求。 非线性 Hartree–Fock 方程的解也表现得好像每个粒子都受到所有其他粒子产生的平均场的影响(参见下面的 Fock 算子),因此术语继续存在。 尽管不动点迭代算法并不总是收敛,但方程组几乎普遍采用迭代法求解。这种求解方案不是xxx可能的方案,也不是哈特里-福克方程序的本质特征。
哈特里-福克方程的典型应用是求解原子、分子、纳米结构和固体的薛定谔方程,但它在核物理中也得到广泛应用。 (有关其在核结构理论中的应用的讨论,请参见 Hartree–Fock–Bogoliubov 方法)。 在原子结构理论中,计算可能针对具有许多激发能级的光谱,因此原子的哈特里-福克方程假设波函数是具有明确定义的量子数的单一配置状态函数,并且能级是 不一定是基态。
对于原子和分子,Hartree–Fock 解决方案是大多数更准确地描述多电子系统的方法的中心起点。
本文的其余部分将着重于适用于以原子为特例的分子的电子结构理论的应用。这里的讨论仅针对受限的哈特里-福克方程,其中原子或分子是一个闭壳层 所有轨道(原子或分子)都被双重占据的系统。 一些电子未配对的开壳层系统可以通过受限开壳层或不受限哈特里-福克方程序处理。
简史
编辑早期的半经验方法
哈特里-福克方程的起源可以追溯到 20 年代末,即 1926 年薛定谔方程发现后不久。 1920 年代(E. Fues、R. B. Lindsay 和他自己)以玻尔的旧量子理论为背景。
在原子的玻尔模型中,主量子数为 n 的态的能量以原子单位表示为 E = − 1 / n 2 {displaystyle E=-1/n{2}} 。 从原子光谱中观察到,多电子原子的能级可以通过应用玻尔公式的修改版本得到很好的描述。 通过引入量子缺陷 d 作为经验参数,一般原子的能级可以用公式 E = − 1 / ( n + d ) 2 {displaystyle E=-1/(n+d){ 2}} ,从某种意义上说,人们可以很好地再现在 X 射线区域观察到的跃迁水平(例如,参见莫斯利定律中的经验讨论和推导)。 非零量子缺陷的存在归因于电子-电子排斥,这在孤立的氢原子中显然不存在。
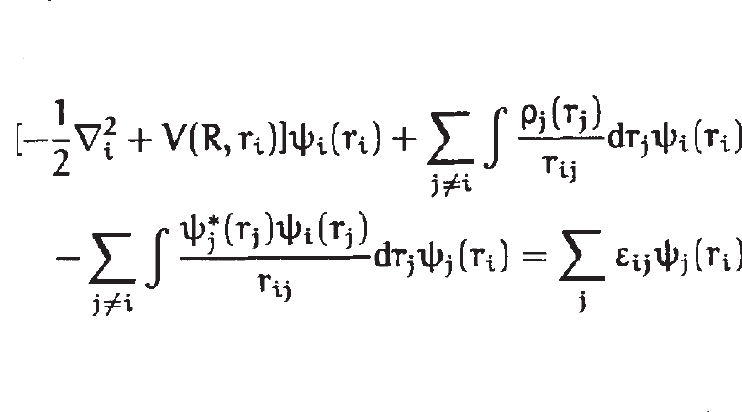

这种排斥导致部分屏蔽裸露的核电荷。 这些早期研究人员后来引入了包含额外经验参数的其他势,希望能更好地再现实验数据。
内容由匿名用户提供,本内容不代表vibaike.com立场,内容投诉举报请联系vibaike.com客服。如若转载,请注明出处:https://vibaike.com/220353/