
重症肌无力
编辑重症肌无力(myasthenia gravis,MG)是一种主要由于神经-肌肉接头突触后膜上乙酰胆碱受体(AChR)受损引起的神经-肌肉接头传递功能障碍的获得性自身免疫性疾病。临床主要表现为部分或全身骨骼肌无力和极易疲劳,活动后症状加重,经休息和胆碱酯酶抑制剂治疗后症状减轻。
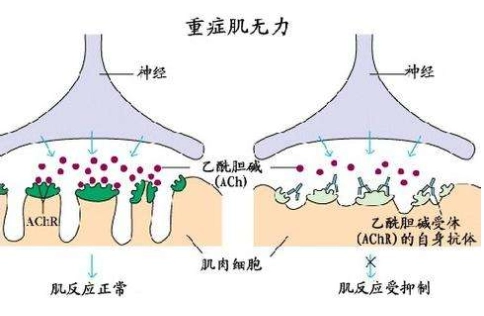
<SPan style=”color: #262626;font-family: var(–theme-font-family)”>神经肌肉接头由突触前膜、突触间隙和突触后膜构成。神经末梢合成乙酰胆碱,通过突触囊泡释放入突触间隙。突触后膜上有许多褶皱,乙酰胆碱受体分布在这些褶皱的嵴上。乙酰胆碱与乙酰胆碱受体结合后,终板上阳离子通道开放,产生终板电位,引起肌纤维收缩。
重症肌无力,属于一组神经系统疾病。其特征是神经和肌肉之间的信号传输受到干扰,导致肌肉高度易疲劳,并被概括为冲动的神经肌肉传输障碍或肌无力综合征。
它是一种非遗传性自身免疫性疾病,其中横纹肌(骨骼肌)的运动终板出现紊乱,其原因尚未完全研究,也包括单块或多块肌肉,无论一侧身体。
主要症状包括它们不断变化的特征,例如,除了受影响的肌肉发生变化外,它们通常会在一天中无缘无故地自发增加,和/或受影响的肌肉在休息时会突然恢复。
重症肌无力发生在人和动物身上,尤其是家犬。
人类重症肌无力的频率
编辑重症肌无力是一种比较少见的疾病。疾病频率(患病率)约为每 100 万居民 100 至 200 种疾病。本病可在任何年龄发病,但有两个发病高峰。
第 一个高峰是在生命的第二个和第三个十年之间,偏好女性,第二个高峰在生命的第六个和第八个十年之间,偏好男性。
重症肌无力在女性中更为常见(比例3:2)。
自 1950 年左右开始流行病学调查以来,该病的发病率有所增加;在1990年代,发病率大约高出四倍。这种增长归因于对该疾病的认识提高、诊断方法改进、死亡率降低以及人口年龄结构发生变化。
发病年龄分类
编辑按发病年龄分型
按发病年龄及临床表现将重症肌无力分为成年型、儿童型、少年型。
成年型(Osserman分型)
根据临床表现及受累肌群,成年型可分为I眼肌型、IIA轻度全身型、IIB中度全身型、III急性重症型、IV迟发重症型、V肌萎缩型6型。
- I眼肌型(15%~20%):病变仅限于眼外肌,出现上睑下垂和复视。
- IIA轻度全身型(30%):可累及眼、面、四肢肌肉,生活多可自理,无明显咽喉肌受累。
- IIB中度全身型(25%):四肢肌群受累明显,眼外肌麻痹,较明显的咽喉肌无力症状,如说话含糊不清吞咽困难、饮水呛咳、咀嚼无力,但呼吸肌受累不明显。
- III急性重症型(15%):急性起病,常在数周内累及延髓肌、肢带肌、躯干肌和呼吸肌,肌无力严重,有重症肌无力危象,需做气管切开,死亡率较高。
- IV迟发重症型(10%):病程达2年以上,常由I、IIA、IIB型发展而来,症状同III型,常合并胸腺瘤,预后较差。
- V肌萎缩型:少数患者肌无力伴肌萎缩。
儿童型
多数仅限于眼外肌麻痹,双眼睑下垂可交替出现呈拉锯状,约1/4病例可自然缓解,约1/4病例累及全身骨骼肌,仅少数病例累及全身骨骼肌。分为新生儿型、先天性肌无力综合征。
- 新生儿型:MG孕妇将AChR抗体IgG经胎盘传给胎儿,患儿出生后即哭声低、吸吮无力、肌张力低动作减少。
- 先天性肌无力综合征:出生后短期内出现持续的眼外肌麻痹,常有阳性家族史,但其母亲未患MG。
少年型
多在10岁后发病,多为单纯眼外肌麻痹,部分伴吞咽困难及四肢无力。
病因及发展
编辑重症肌无力是一种自身免疫性疾病,这意味着身体会形成针对身体自身结构的自身抗体。在重症肌无力中,抗体针对神经肌肉接头区域的突触后膜结构。到目前为止,最常见的是乙酰胆碱受体抗体,在大约 85% 的病例中,即针对烟碱型乙酰胆碱受体的抗体。
在 1% 到 10% 的受影响者中,可以检测到针对肌肉特异性酪氨酸激酶 (MuSK) 的抗体,在一些受影响的人中可以检测到所谓的低亲和力乙酰胆碱受体抗体或针对脂蛋白受体相关蛋白 (LRP4) 的抗体。
在一些极有可能患有重症肌无力病的患者中,无法检测到抗体(血清阴性重症肌无力)。
据推测,除了提到的抗体之外,可能还有其他与肌无力相关的抗体。只有与肌无力相关抗体和胸腺的现有联系以及该疾病的因果基础,即神经和肌肉之间的干扰信号传输,已得到证实。环境影响、感染、炎症、精神和心理压力等波动症状的触发因素也不清楚。
乙酰胆碱受体抗体
乙酰胆碱受体抗体阻止或增加递质乙酰胆碱与其受体之间的相互作用。因此,电脉冲(动作电位)不能再从神经传递到肌肉,肌肉也就不兴奋了。
此外,乙酰胆碱受体的数量减少,因为抗体与乙酰胆碱受体的结合会通过免疫活性降解它们。亚突触膜的结构分解成碎片。自噬体通过内吞作用形成。带有消化酶的运输囊泡与自噬体融合。
由于这种免疫反应,乙酰胆碱受体每两到三天就会降解一次。改变了电机端板的结构。亚突触连接褶皱变平,突触间隙变宽。
结果,乙酰胆碱在释放时扩散出突触间隙,或者在它占据乙酰胆碱受体之前被胆碱酯酶水解。
胸腺的变化
在重症肌无力的情况下,通常可以检测到胸腺的病理变化。胸腺在自身免疫过程的发展中起着至关重要的作用。
在高达 70% 的病例中,具有活跃生发中心的胸腺炎(淋巴滤泡增生)很明显。在 10-15% 的受影响者中可以检测到胸腺瘤。
在某些情况下,手术切除胸腺可对疾病进程产生积极影响。 任何年龄都应有手术指征。
临床表现
早上和休息后的表现最 好,但经过几次重复运动后,各个肌肉或整个肌肉群都会变得疲惫不堪。晚上症状通常更严重。小肌肉尤其会受到麻痹的影响或最初仅受限的活动性,但原则上所有横纹(自愿活动)的肌肉都会受到影响。
没有运动终板的肌肉组织,如心肌和平滑肌,不受这种疾病的影响。然而,科学上从未排除后者的参与。
他们参与的线索可能是基于吡啶斯的明的肌无力药物对肠道活动的有效性,因为肠道(除了随意括约肌)由平滑肌组成。
另一个迹象可能是与肠道有关的便秘,这是兰伯特-伊顿-鲁克综合征 (LEMS) 的症状之一,也被怀疑是由自身免疫系统引起的。
在大约 50% 的病例中,重症肌无力首先在眼睛、眼睑和/或外眼部肌肉中变得明显:眼睑肌肉疲劳会导致典型的“卧室视图”(上睑下垂),因为眼睑无法再抬起,患者将头向后仰以查看眼睑下方。
随着疾病的进展,在大多数情况下,手臂比腿部受到的影响更严重。此外,呼吸肌可能会严重受损,以至于受影响的人只能坐着睡觉,或者在晚期阶段必须通风。吞咽肌肉也可能受到如此严重的影响,以至于需要通过胃管进行支持或全面护理。
平衡、退行性记忆或感觉障碍不是重症肌无力的征兆。
至于症状的特征,应该说它们是在变化的,而不是静止和僵硬的;在时间、负荷的触发强度和当前受影响的肌肉或肌肉群的“选择”方面都在变化——例如左手右眼,一次一起,一次单独或早上或晚上。
症状不可预测,通常突然出现。 一些肌肉或肌肉群仅在活动方面受到限制,而其他人可能根本没有足够的力量来移动它们。
由于病程的这种可变性在受影响的个体或患者与患者之间存在差异,因此不能真正谈论一般的临床表现(外观)。
内容由匿名用户提供,本内容不代表vibaike.com立场,内容投诉举报请联系vibaike.com客服。如若转载,请注明出处:https://vibaike.com/337025/