
酶抑制剂
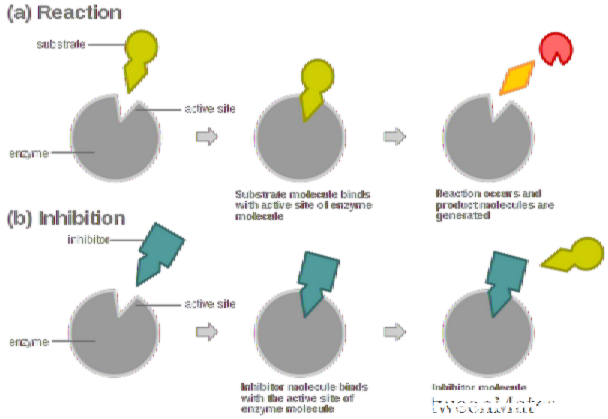
编辑酶抑制剂是一种分子结合于酶并降低它的活性。通过与酶的活性位点结合,抑制剂降低了底物与酶的相容性,从而抑制了酶-底物复合物的形成,阻止了反应的催化作用,并减少了(有时为零)反应。可以说,随着酶抑制剂浓度的增加,酶活性的速率降低,因此,产物的产生量与抑制剂分子的浓度成反比。由于阻断酶的活性可以杀死病原体或纠正代谢失衡,许多药物是酶抑制剂。它们还用于杀虫剂中。并非所有与酶结合的分子都是抑制剂。酶激活剂与酶结合并增加其酶促活性,而酶底物在酶的正常催化循环中结合并转化为产物。
抑制剂的结合可以阻止底物进入酶的活性位点和/或阻止酶催化其反应。抑制剂结合是可逆的或不可逆的。不可逆抑制剂通常会与酶反应并进行化学改变(例如,通过共价键形成)。这些抑制剂修饰了酶促活性所需的关键氨基酸残基。相反,可逆抑制剂非共价结合,并且取决于这些抑制剂是否结合酶,酶-底物复合物或两者结合而产生不同类型的抑制。
许多药物分子是酶抑制剂,因此它们的发现和改进是生物化学和药理学研究的活跃领域。药用酶抑制剂通常由其特异性(缺乏与其他蛋白质的结合)和效力(其解离常数,表示抑制该酶所需的浓度)来判断。高的特异性和效力确保药物几乎没有副作用,因此毒性低。
酶抑制剂也天然存在,并参与代谢的调节。例如,代谢途径中的酶可以被下游产物抑制。这种类型的负反馈减慢生产线时,产品开始建立并保持的重要途径平衡的细胞。其他细胞酶抑制剂是特异性结合并抑制酶靶标的蛋白质。这可以帮助控制可能损害细胞的酶,例如蛋白酶或核酸酶。一个很好的例子是核糖核酸酶抑制剂,它与核糖核酸酶是已知最紧密的蛋白质-蛋白质相互作用之一。天然酶抑制剂也可以是有毒物质,可用作防御捕食者或杀死猎物的方法。
可逆抑制剂的例子
编辑由于酶已经进化为紧密结合其底物,并且大多数可逆抑制剂都结合在酶的活性位点上,因此,其中一些抑制剂的结构与目标底物的结构惊人相似就不足为奇了。DHFR的抑制剂是突出的例子。这些底物模拟物的另一个例子是蛋白酶抑制剂,这是一类非常成功的用于治疗HIV的抗逆转录病毒药物。利托那韦的结构,利托那韦是一种基于肽并含有三个肽键的蛋白酶抑制剂,显示在右侧。由于这种药物类似于HIV蛋白酶底物的蛋白质,因此它在酶的活性位点与该底物竞争。
通常将酶抑制剂设计为模拟酶催化反应的过渡态或中间体。这确保了抑制剂利用了酶的过渡态稳定作用,从而导致了比基于底物的设计更好的结合亲和力(更低的K i)。这种过渡状态抑制剂的一个例子是抗病毒药奥司他韦; 这种药物在病毒酶神经氨酸酶的反应中模仿了环氧鎓离子的平面性质。
但是,并非所有抑制剂都基于底物的结构。例如,另一种HIV蛋白酶抑制剂替普拉那韦的结构显示在左侧。该分子不是基于肽,并且与蛋白质底物没有明显的结构相似性。这些非肽抑制剂可能比含有肽键的抑制剂更稳定,因为它们不是肽酶的底物,并且降解的可能性较小。
在药物设计中,重要的是要考虑目标酶所暴露的底物浓度。例如,某些蛋白激酶抑制剂的化学结构类似于三磷酸腺苷,这些酶的底物之一。但是,作为简单竞争性抑制剂的药物必须与细胞中高浓度的ATP竞争。蛋白激酶还可以通过在激酶与其底物蛋白相互作用的结合位点竞争而受到抑制,并且大多数蛋白以远远低于ATP浓度的浓度存在于细胞内。结果,如果两种蛋白激酶抑制剂都以相似的亲和力结合在活性位点上,但是只有一种必须与ATP竞争,那么蛋白结合位点上的竞争性抑制剂将更有效地抑制酶。
抑制剂的发现和设计
编辑新药是漫长的药物开发过程的产物,第一步通常是发现新的酶抑制剂。过去,发现这些新抑制剂的唯一方法是反复试验:针对目标酶筛选庞大的化合物文库,并希望能出现一些有用的线索。这种蛮力方法仍然是成功的,甚至通过组合化学方法得以扩展,该组合化学方法可快速产生大量新型化合物和高通量筛选技术,以快速筛选这些庞大的化学文库中有用的抑制剂。
最近,一种替代方法被应用:合理的药物设计使用酶活性位点的三维结构来预测哪些分子可能是抑制剂。然后测试了这些预测,这些测试的化合物之一可能是新型抑制剂。然后使用这种新的抑制剂尝试获得抑制剂/酶复合物中酶的结构,以显示分子如何与活性位点结合,从而允许对抑制剂进行更改以尝试优化结合。然后重复该测试和改善周期,直到产生足够有效的抑制剂。基于计算机的方法预测抑制剂对酶的亲和力的方法也在开发中,例如分子对接和分子力学。
抑制剂的用途
编辑酶抑制剂存在于自然界中,也作为药理学和生物化学的一部分进行设计和生产。天然毒物通常是酶抑制剂,可以进化来保护植物或动物免受食肉动物侵害。这些天然毒素包括一些已知最剧毒的化合物。人工抑制剂通常用作药物,但也可以是杀虫剂(如马拉硫磷)、除草剂(如草甘膦)或消毒剂(如三氯生)。其他人工酶抑制剂可阻断乙酰胆碱酯酶(一种分解酶)乙酰胆碱,在化学战中用作神经毒剂。
化学疗法
编辑酶抑制剂的最常见用途是作为治疗疾病的药物。这些抑制剂中有许多靶向人的酶并旨在纠正病理状况。但是,并非所有药物都是酶抑制剂。有些药物(例如抗癫痫药)通过引起或多或少的酶的产生来改变酶的活性。这些作用称为酶诱导和抑制,是基因表达的改变,与本文讨论的酶抑制类型无关。其他药物与不是酶的细胞靶标相互作用,例如离子通道或膜受体。
在比较甲氨蝶呤与叶酸的药物中,一些抑制剂与它们靶向的酶的底物结构相似的另一个例子。叶酸是二氢叶酸还原酶的底物,二氢叶酸还原酶是一种参与制造核苷酸的酶,被甲氨蝶呤有效抑制。甲氨蝶呤阻断了二氢叶酸还原酶的作用,从而停止了核苷酸的产生。这种核苷酸的生物合成阻滞比非分裂细胞对快速生长的细胞更具毒性,因为快速生长的细胞必须进行DNA复制,因此甲氨蝶呤常用于癌症化疗。
抗生素
编辑药物还用于抑制病原体生存所需的酶。例如,细菌被厚的细胞壁包围,该细胞壁由称为肽聚糖的网状聚合物制成。许多抗生素(例如青霉素和万古霉素)会抑制产生这种酶的酶,然后将这种聚合物的链交联在一起。这会导致细胞壁强度下降,细菌破裂。在该图中,显示了青霉素分子(以球形和棒状显示)与其靶标结合,即细菌Streptomyces R61的转肽酶(该蛋白显示为带状图)。
当人体中缺少病原体生存所必需的酶或两者之间存在很大差异时,就会促进抗生素药物的设计。在上面的示例中,人类没有制造肽聚糖,因此该过程的抑制剂对细菌具有选择性毒性。利用细菌中核糖体的结构差异或它们如何产生脂肪酸,在抗生素中也会产生选择性毒性。
代谢控制
编辑酶抑制剂在代谢控制中也很重要。细胞中的许多代谢途径被代谢物抑制,这些代谢物通过变构调节或底物抑制来控制酶的活性。一个很好的例子是糖酵解途径的变构调节。这种分解代谢途径消耗葡萄糖并产生ATP、NADH和丙酮酸。调节糖酵解的关键步骤是磷酸果糖激酶-1催化的途径中的早期反应(PFK1)。当ATP水平升高时,ATP会结合PFK1中的变构位点,从而降低酶反应的速率。糖酵解受到抑制,ATP产量下降。这种负反馈控制有助于维持细胞中ATP的稳定浓度。然而,代谢途径不仅仅通过抑制来调节,因为酶激活同样重要。对于PFK1、果糖2,6-二磷酸和ADP是变构活化剂的代谢产物。
生理酶抑制作用也可以通过特定的蛋白质抑制剂产生。这种机制发生在胰腺中、胰腺合成了许多被称为酶原的消化前体酶。这些中的许多被胰蛋白酶蛋白酶激活,因此重要的是抑制胰腺中胰蛋白酶的活性以防止器官自身消化。控制胰蛋白酶活性的一种方法是在胰腺中产生特异性和有效的胰蛋白酶抑制剂蛋白。该抑制剂与胰蛋白酶紧密结合,防止了胰蛋白酶的活性,否则会损害器官。尽管胰蛋白酶抑制剂是一种蛋白质,但通过从胰蛋白酶的活性位点排除水并破坏过渡态的稳定性,可以避免被蛋白酶水解为底物。生理酶抑制剂蛋白的其他实例包括细菌核糖核酸酶barnase的barstar抑制剂。
农药
编辑许多农药是酶抑制剂。乙酰胆碱酯酶(AChE)是一种在动物中发现的酶,从昆虫到人类。它通过将神经递质乙酰胆碱分解成其成分乙酸盐和胆碱的机制,对神经细胞功能至关重要。这在神经递质中有些不同寻常,因为包括5-羟色胺,多巴胺和去甲肾上腺素在内的大多数神经递质都从突触间隙吸收而不是裂解。大量的AChE抑制剂用于医学和农业领域。可逆竞争性抑制剂,例如edrophonium,毒扁豆碱和新斯的明用于重症肌无力的治疗和麻醉。的氨基甲酸酯杀虫剂也可逆AChE抑制物的例子。在有机磷农药如马拉硫磷,对硫磷和毒死蜱不可逆地抑制乙酰胆碱酯酶。
草甘膦除草剂是3-磷酸钾的1-羧基乙烯基转移酶的抑制剂,其他除草剂,例如磺酰脲类抑制乙酰乳酸合酶。这两种酶都是植物制造支链氨基酸所必需的。除草剂还可以抑制许多其他酶,包括脂质和类胡萝卜素的生物合成以及光合作用和氧化磷酸化过程所需的酶。
内容由匿名用户提供,本内容不代表vibaike.com立场,内容投诉举报请联系vibaike.com客服。如若转载,请注明出处:https://vibaike.com/116459/